
Kako nam je u ordinaciji nekoliko pacijenata imalo genetički potvrđen Joubert sindrom, smatrali smo da je interesantno opisati sa više detalja ovaj sindrom.
Joubert syndrome je retka genetička bolest koju karakteriše nepravilan razvoj pojedinih delova mozga a koji se karakteriše problemima u intelektualnom i motorickom razvoju , bubrežnoj i jetrinoj funkciji kao i očnim poremećajima.
Nasleđuje se autozomno recesivno i najvise pogađa strukture mozga zadužene za balans i koordinaciju koji zovemo mali mozak (cerebellum). Iz tog razloga su najupečatljiviji simptomi hipotonija, odsustvo kontrole mišića – ataksija, problem sa disanjem (Hiperpnea), sleep apnea, abnormalni pokreti očiju i nemogućnost kretanja očnih jabučica u svim pravcima (okulomotorna dispraksija), epileptični napadi.
Jouber sindrom je prvi put opisala Dr Marie Joubert, dečiji neurolog 1969.god. kada je sa svojim kolegama opisala 4 člana porodice koji su imali nerazvijen malomoždani crv – deo koji spaja dve malomoždane hemisfere (vermis) a imali simptome pojačane respiracije (hiperpnea), nenormalne pokrete očiju, ataxiju i intelektualnu nedovoljnost. Nekoliko godina kasnije sa razvojem neuroimaging tehnika pronađen je specifični neuroradiološki nalaz – “znak molara” koje je postao važan dijagnostički krirerijum za dijagnozu JS.
S obzirom da zahvata više organa (oči, jetra, bubreg, mozak, skelet) spada u multiorganske poremećaje. Među druge probleme sa očima koji se mogu naći kod Joubert sindroma spadaju: nenormalnosti mrežnjače, poigravanje očiju (nystagmus), razrokost (strabizam), poremećaj dužice (kolobom) ili pad kapka (ptoza). Takođe mogu biti priduženi poremećaji skeletnog Sistema u vidu 6. prsta (polidaktilija) i na nogama i na rukama, hormonski poremećaji, rascep mekog nepca, malformacije jezika, koortikomedularne ciste bubrega, policistični bubrezi, i fibroza jetre kao i poremećaj na nivou moždanih ovojnica u vidu encefalocela, kao i epileptične napade.
Na magnetnoj rezonanci mozga (MRI brain) koja je veoma vazna za neuroimaging dijagnozu Joubert sindroma postoji tipičan znak molara (“molar tooth sign”) koga karakteriše odsustvo ili nedovoljno razvijeni malomoždani vermis i nenormalnosti moždanog stabla.
Od tada je razvojem genetike pronađeno vise od 35 gena koji su zaduženi za kliničko ispoljavanje Joubert sindroma. Molekularna dijagnoza JS moze biti postavljena u 60-90% slučajeva.
U zavisnosti od zahvaćenog gena zavisi i klinička prezentacija Joubert sindroma. Mutacija AHI1 (JBTS3) gena javlja se u oko ~7%-10% porodica. Obolele osobe sa ovom mutacijom imaju problem sa vidom zbog retinalne distrofije. Mutacija NPHP1 (JBTS4) gena uzrokuje oko 1-2% JS. Osobe sa ovom mutacijom imaju progresivnu bubrežnu boest. Mutacija CEP290 (JBTS5) gena odgovorna je za oko 7-10% of JS. Mutacije gena CPLANE1, CC2D2A, INPP5E, KIAA0586, MKS1, RPGRIP1L TCTN2, TMEM67 and TMEM216 su ređe ali su takođe odgovorne za JS.
Retki slučajevi JS uzrokovni su mutacijom OFD1 gena koji se nasleđuje recesivno vezano za X hromozom i najčešće se javlja kod muškog pola. Ženski pol koji nosi mutaciju je samo nosilac patološkog gena (Carrier ) i ne ispoljava kliničke simptome jer ima dva X fromozoma dok dečaci imaju samo 1 pa je ispoljavanje bolesti vezano za X hromozom neizbezno.
Dijagnostika i praćenje osoba sa JS
Već smo napomenuli da je JS multisistemsko oboljenje te shodno tome dijagnostika i praćenja obuhvata neurološku genetsku i pedijatrijsku oblast.
Zbog toga je za dijagnozu i dalje praćenje osoba sa JB potrebno:
- Neuroradiološko ispitivanje – MRI mozga radi otkrivanja moždanih malformacija (“molar tooth sign”)
- Bazični neuološki pregled tonusa, motorike, očnih pokreta , respiracija i psihomotornog razvoja
- EEG ukoliko pacijent s JS ima epileptične napade
- Konsultacija oftalmologa zbog očnih abnormalnosti (retinalne distrofije, strabizma i koloboma)
- Ultrazvučni pregled abdomena (praćenje funkcije jetre i bubrega)
- Laboratorijske analize uključujući krvnu sliku, renalnu i jetrinu funkciju, urea i kreatinin, analiza urina
- Radiografija ruku i nogu u slučaju dodatnog prsta na rukama ili nogama
- Konsultacija genetičara radi mogućeg genetskog testa i genetskog savetovanja
Tretman JS
Tretman JS je obično simptomatski i supurativan. Usporen psihomotorni razvoj zahteva intenzivan program psihomotorne reedukacije sa timom stručnjaka kao što su logoped, defektolog, fizioterapeut, okupaciona terapija, nutricionista.
Multidisciplinarni pristup je neophodan uključujući sledeće vrste specijalnosti: dečjeg neurologa, fizijatra, oftalmologa, dečjeg nefrologa i gastroenterologa, genetičara. Praćenje funkcije bubrega i jetre treba obavljati na godišnjem nivou.
Uloga neurologa je naročito značajna u slučajevima JS koji imaju epileptične napade.
Više o epileptičnim napadima pročitajte ovde: Tipovi epileptičnih napada – Neuro Kid Bossa
Postoji udruženje pacijenata sa JS i njihovih familija koje je počelo sa radom 1992. godine i detalje o njihovom radu i borbi mozete naći na sajtu: https://jsrdf.org/
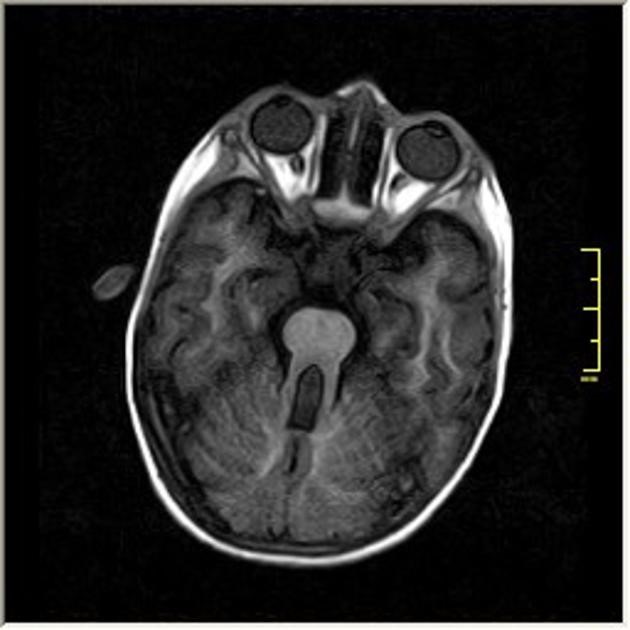
Karakterističan molar tooth sign na MRI mozga (izgled zuba-molara)
.
Get Your Molar Tooth Right: Joubert Syndrome Misdiagnosis Unmasked by Whole-Exome Sequencing.
D’Abrusco F, Arrigoni F, Serpieri V, Romaniello R, Caputi C, Manti F, Jocic-Jakubi B, Lucarelli E, Panzeri E, Bonaglia MC, Chiapparini L, Pichiecchio A, Pinelli L, Righini A, Leuzzi V, Borgatti R, Valente EM.Cerebellum. 2022 Dec;21(6):1144-1150. doi: 10.1007/s12311-021-01350-8. Epub 2021 Nov 30.PMID: 34846692
Phenotypic spectrum and prevalence of INPP5E mutations in Joubert syndrome and related disorders.
Travaglini L, Brancati F, Silhavy J, Iannicelli M, Nickerson E, Elkhartoufi N, Scott E, Spencer E, Gabriel S, Thomas S, Ben-Zeev B, Bertini E, Boltshauser E, Chaouch M, Cilio MR, de Jong MM, Kayserili H, Ogur G, Poretti A, Signorini S, Uziel G, Zaki MS; International JSRD Study Group; Johnson C, Attié-Bitach T, Gleeson JG, Valente EM.Eur J Hum Genet. 2013 Oct;21(10):1074-8. doi: 10.1038/ejhg.2012.305. Epub 2013 Feb 6.PMID: 23386033 Free PMC article.
Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies.
Iannicelli M, Brancati F, Mougou-Zerelli S, Mazzotta A, Thomas S, Elkhartoufi N, Travaglini L, Gomes C, ArdissinoGL, Bertini E, Boltshauser E, Castorina P, D’Arrigo S, FischettoR, Leroy B, Loget P, Bonnière M, Starck L, Tantau J, GentilinB, Majore S, Swistun D, Flori E, Lalatta F, Pantaleoni C, Penzien J, Grammatico P; International JSRD Study Group; Dallapiccola B, Gleeson JG, Attie-Bitach T, Valente EM.HumMutat. 2010 May;31(5):E1319-31. doi: 10.1002/humu.21239.PMID: 20232449 Free PMC article.