
As several patients in our outpatient clinic had genetically confirmed Joubert syndrome, we thought it would be interesting to describe this syndrome in more detail.
Joubert syndrome is a rare genetic disease characterized by irregular development of certain parts of the brain, characterized by problems in intellectual and motor development, kidney and liver function, as well as eye disorders.
It is inherited in an autosomal recessive manner and mostly affects the brain structures responsible for balance and coordination, which we call the cerebellum. For this reason, the most striking symptoms are hypotonia, lack of muscle control - ataxia, breathing problems (Hyperpnea), sleep apnea, abnormal eye movements and the inability to move the eyeballs in all directions (oculomotor dyspraxia), epileptic seizures.
Joubert syndrome was first described by Dr. Marie Joubert, a child neurologist in 1969. when she and her colleagues described 4 family members who had an underdeveloped cerebellar vermis - the part that connects the two cerebellar hemispheres (vermis) and had symptoms of increased respiration (hyperpnea), abnormal eye movements, ataxia and intellectual disability. A few years later, with the development of neuroimaging techniques, a specific neuroradiological finding was found - the "molar sign" which became an important diagnostic criterion for the diagnosis of JS.
Given that it affects several organs (eyes, liver, kidney, brain, skeleton), it belongs to multiorgan disorders. Other eye problems that can be found in Joubert syndrome include: retinal abnormalities, eye twitching (nystagmus), wide-eyed vision (strabismus), iris disorder (coloboma), or eyelid droop (ptosis). There can also be associated disorders of the skeletal system in the form of the 6th finger (polydactyly) both on the legs and on the hands, hormonal disorders, cleft soft palate, tongue malformations, corticomedullary kidney cysts, polycystic kidneys, and liver fibrosis as well as a disorder at the level of the meninges in such as encephalocele, as well as epileptic seizures.
On magnetic resonance imaging of the brain (MRI brain), which is very important for the neuroimaging diagnosis of Joubert syndrome, there is a typical molar tooth sign characterized by the absence or insufficiently developed cerebellar vermis and brainstem abnormalities.
Since then, with the development of genetics, more than 35 genes have been found that are responsible for the clinical manifestation of Joubert syndrome. Molecular diagnosis of JS can be established in 60-90% of cases.
The clinical presentation of Joubert syndrome also depends on the affected gene. Mutation AHI1 (JBTS3) gena javlja se u oko ~7%-10% porodica. Obolele osobe sa ovom mutacijom imaju problem sa vidom zbog retinalne distrofije. Mutacija NPHP1 (JBTS4) gena uzrokuje oko 1-2% JS. Osobe sa ovom mutacijom imaju progresivnu bubrežnu boest. Mutacija CEP290 (JBTS5) gene is responsible for about 7-10% of JS. Gene mutations CPLANE1, CC2D2A, INPP5E, KIAA0586, MKS1, RPGRIP1L TCTN2, TMEM67 and TMEM216 are less common but are also responsible for JS.
Rare cases of JS are caused by a mutation OFD1 gene that is inherited recessively linked to the X chromosome and most often occurs in males. Females who carry the mutation are only carriers of the pathological gene (Carrier) and do not show clinical symptoms because they have two X chromosomes, while boys have only 1, so the manifestation of the disease related to the X chromosome is inevitable.
Diagnostics and follow-up of patients with JS
We have already mentioned that JS is a multisystem disease, and accordingly, diagnosis and follow-up includes the neurological, genetic and pediatric areas.
Therefore, for the diagnosis and further follow-up of people with JB, it is necessary:
- Neuroradiological examination - MRI of the brain to detect brain malformations ("molar tooth sign")
- Basic neurological examination of tone, motor skills, eye movements, respiration and psychomotor development
- EEG if the JS patient has epileptic seizures
- Ophthalmologist consultation for eye abnormalities (retinal dystrophy, strabismus and coloboma)
- Ultrasound examination of the abdomen (monitoring of liver and kidney function)
- Laboratory tests including blood count, renal and liver function, urea and creatinine, urine analysis
- X-ray of hands and feet in case of extra finger or toe
- Consultation of a geneticist for a possible genetic test and genetic counseling
Treatment of JS
Treatment of JS is usually symptomatic and suppurative. Slowed psychomotor development requires an intensive program of psychomotor reeducation with a team of experts such as a speech therapist, a speech therapist, a speech therapist, a physiotherapist, occupational therapy, and a nutritionist.
A multidisciplinary approach is necessary including the following types of specialties: pediatric neurologist, physiatrist, ophthalmologist, pediatric nephrologist and gastroenterologist, geneticist. Monitoring of kidney and liver function should be done on an annual basis.
The role of the neurologist is particularly important in cases of JS who have epileptic seizures.
Read more about epileptic seizures here: Types of Epileptic Seizures - Neuro Kid Bossa
There is an association of patients with JS and their families that started working in 1992 and you can find details about their work and struggle on the website: https://jsrdf.org/
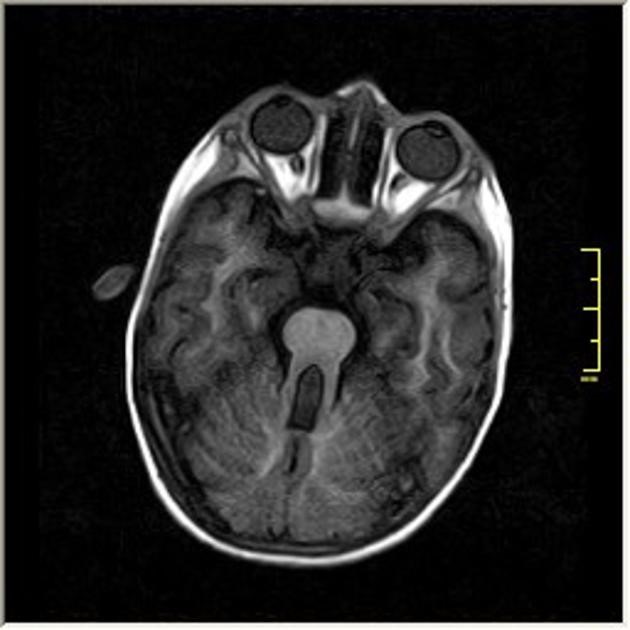
Characteristic molar tooth sign on brain MRI (tooth-molar appearance)
.
Get Your Molar Tooth Right: Joubert Syndrome Misdiagnosis Unmasked by Whole-Exome Sequencing.
D’Abrusco F, Arrigoni F, Serpieri V, Romaniello R, Caputi C, Manti F, Jocic-Jakubi B, Lucarelli E, Panzeri E, Bonaglia MC, Chiapparini L, Pichiecchio A, Pinelli L, Righini A, Leuzzi V, Borgatti R, Valente EM.Cerebellum. 2022 Dec;21(6):1144-1150. doi: 10.1007/s12311-021-01350-8. Epub 2021 Nov 30.PMID: 34846692
Phenotypic spectrum and prevalence of INPP5E mutations in Joubert syndrome and related disorders.
Travaglini L, Brancati F, Silhavy J, Iannicelli M, Nickerson E, Elkhartoufi N, Scott E, Spencer E, Gabriel S, Thomas S, Ben-Zeev B, Bertini E, Boltshauser E, Chaouch M, Cilio MR, de Jong MM, Kayserili H, Ogur G, Poretti A, Signorini S, Uziel G, Zaki MS; International JSRD Study Group; Johnson C, Attié-Bitach T, Gleeson JG, Valente EM.Eur J Hum Genet. 2013 Oct;21(10):1074-8. doi: 10.1038/ejhg.2012.305. Epub 2013 Feb 6.PMID: 23386033 Free PMC article.
Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies.
Iannicelli M, Brancati F, Mougou-Zerelli S, Mazzotta A, Thomas S, Elkhartoufi N, Travaglini L, Gomes C, ArdissinoGL, Bertini E, Boltshauser E, Castorina P, D’Arrigo S, FischettoR, Leroy B, Loget P, Bonnière M, Starck L, Tantau J, GentilinB, Majore S, Swistun D, Flori E, Lalatta F, Pantaleoni C, Penzien J, Grammatico P; International JSRD Study Group; Dallapiccola B, Gleeson JG, Attie-Bitach T, Valente EM.HumMutat. 2010 May;31(5):E1319-31. doi: 10.1002/humu.21239.PMID: 20232449 Free PMC article.