
Definicija, prevalenca i incidenca
Teška mioklonička epilepsija detinjstva (Severe Myoclonic Epilepsy in Infancy) predstavlja jednu od razvojnih Epileptičnih Encefalopatija i prvi put je opisana 1978.g u Francuskoj od strane poznatog francuskog epileptologa iz Marseja Prof. Charlotte Dravet po kojoj je i dobila ime – Dravet syndrome (Dravet&Oguni 2013).
Oko 1.4% sve dece sa epilepsijom ima DS. Incidenca je 1/15700 u USA i u opštoj populaciji 1/40900 i DS obuhvata oko 10% svih razvojnih Epileptičkih Encefalopatija.
U poslednjih 10 godina zahvaljujući podizanju svesti o ovoj bolesti među lekarima koji se bave dečjom neurologijom i epileptologijom i rapidnom razvoju genetike i njenoj dostupnosti u velikom broju zemalja, dijagnoza se brže postavlja, što omogućava brže i efikasnije započinjanje medikamentnog tretmana.
Kliničke karakteristike DS mogu se podeliti u 3 faze (Gataullina &Dulac, 2016):
Prva faza tzv. „Febrilna faza“ u prvoj godini života karakteriše se :
- febrilnim unilateralnim kloničkim napadima, ili generalizovanim napadima u stanjima febrilnosti zbog infekcija ili febrilnosti posle vakcinacije ili nakon kupanja u toploj vodi
- febrilnim status epilepticusima (napadima dužim od 5 minuta) koji bivaju zamenjeni unilateralnim afebrilnim napadima, kloničkim ili drugim fokalnim napadima
- Oko 90% dece ima napad u 1. godini života ( od 4.-8. meseca).
- Deca su prethodno potpuno zdrava i nemaju nikakve neurološke ili druge probleme.
Druga faza tzv „Faza pogoršanja“ od 1.-4. godine života karakteriše se:
- kratkim miokloničkim napadima ili masivnim miklonijama
- atipičnim apsansima
- fokalnim motornim napadima sa devijacijom glave i pogleda u jednu stranu
- unilateralnim motornim napadima koji su kraćeg trajanja nego u predhodnoj fazi
- epizodama padanja glave i produženim fazama „zurenja u prazno“
- fokalnim autonomnim napadima praćenim crvenilom, bledilom ili cijanozom
- kognitivni pad postaje vidljiv u ovoj fazi
- napadi mogu biti trigerovani umorom ili uzbuđenjem, manje su osetljivi na povišenje temperature
Treća faza tzv. „Faza stabilizacije“ posle 5. godine života, koju karakterišu:
- mioklonije
- napadi su pretežno u spavanju a apsansni napadi mogu nestati
- fokalni napdi se mogu smanjiti brojčano ili perzistirati
- ako se do tada nije razvio ataxičan hod biće vidljiv, kao i poremećaj pokreta
- kognitivni pad koji se uglavnom razvija u 2.ili 3. godini života postaje upadljiviji
Dijagnoza i differencijalna dijagnoza
Već smo više puta napominjali da je dijagnoza epilepsije klinička ali da nam EEG mnogo pomaže u odvajanju fokalnih od generalizovanih tipova epilepsija kao i u dijagnostikovanju specifičnih epileptičnih sindroma koji imaju karakterističan EEG crtež (npr: Infantilni spazmi, apsansna epilepsija).
Na žalost kod DS EEG nije od velike pomoći na početku bolesti jer je kod većine dece uredan, tek kod 1/4 se može javiti nespecifično usporenja osnovne aktivnosti. Međutim do 5. godine života kod većine dece će se na EEG-u javiti epileptiformne promene bilo fokalne, generalizovane ili multifokalne (Genton et al, 2011, Speccio et al. 2012). Takođe je fotosenzitivnost prisutna u oko 50 % dece sa DS tokom bilo koje faze bolesti, pa se intermitentna fotostimulacija koju koristimo kao provokativnu metodu tokom EEG, naročito potencira kod dece sa napred navedenim kliničkim karakteristikama.
MRI mozga nam takođe nije od velike koristi jer je uglavnom normalna kod dece sa DS. U malom broju slučajeva tokom kursa bolesti može se naći kortikalna atrofija, hipokampalna skleroza ili nejasna granica između bele i sive mase.
Najvažniju ulogu u dijagnostici pored kliničkih karakteristika napada čini genetska analiza jer više od 80% sve dece sa DS ima mutaciju na nivou natrijumovih kanala (SCN1A mutacija). Oko 90% su mutacije de novo što znači da ne dolaze od roditelja a samo 4-10% mutacija potiču od roditelja.
Ranije otkrivanje bolesti omogućava ranije započinjanje tretmana adekvatnim lekovima (Valproatima, Stiripentolom, Fenfluraminom i dr.) i izbegavanja lekova koji mogu pogoršati napade kod pacijenata sa DS (kao npr. Blokatori natrijumovih kanala kakvi su Crabamazepine, Oxcarbazepine, Phenytoin, Lamotrigine).
Tretman Dravet syndroma
Najbolje je ilustrovan u preporukama experata dečje epileptologije iz 2022 (Wirelli et al.2022)
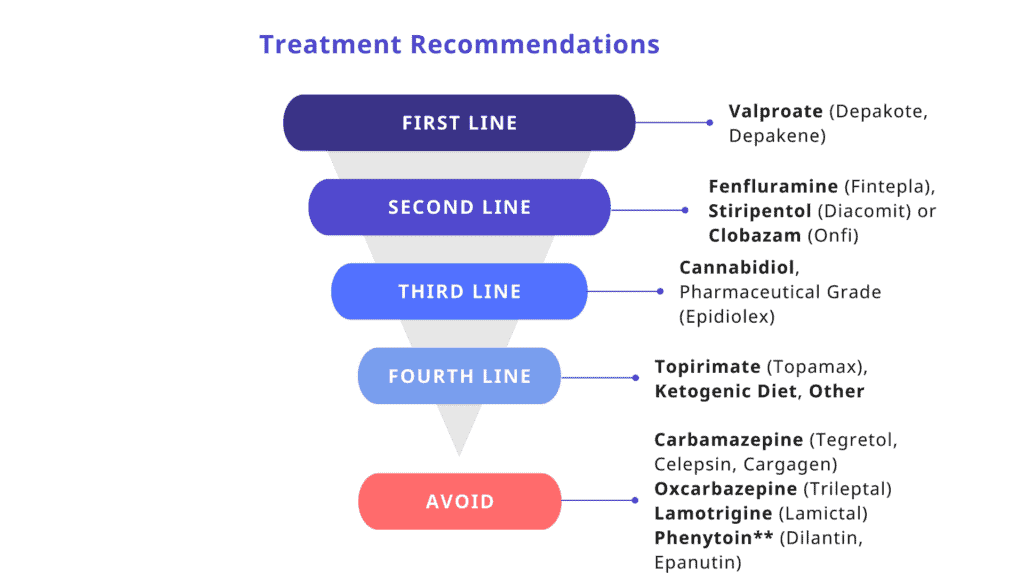
Zbog upadljive fokalnosti napada u 1. Godini života bilo da su unilateralni prolongirani, febrilni ili afebrilni napadi, često se od strane lekara koji nisu familijarni sa ovom dijagnozom ponudi lek Carbamazepine, koji je odličan lek za fokalne napade ali ne i za napade u Dravet sy. Zato je neophodno pre propisivanja bilo kojeg antiepileptičnog leka postaviti najpre dijagnozu vrste napada/Epileptičnog sindroma, a ako to nije moguće u tom trenutku izabrati lek najšireg spektra kakav je Valproat (Eftil) ili Levetiracetam (Keppra). Po postavljanju dijagnoze izabrati lek koji je lek izbora za pomenuti tip epilepsije/Epileptičnog sindroma.
Tok i prognoza
Kako se radi o kompleksnoj epilepsiji, epileptičnoj encefalopatiji koju karakteriše više tipova epileptičnih napada , nemoguće ih je kupirati jednim antiepileptičnim lekom a često i sa kombinacijom nekoliko njih, što onda zovemo farmakorezistencija. U ovom sindromu ne predstavlja problem samo kontrola napada već i sijaset pridružujućih poremećaja kao što su: kognitivne smetnje, poremećaj ponašanja, poremećaj spavanja i hranjenja, tečkoće u učenju i pamćenju, poremećaj hoda i govora, endokrine i autonomne smetnje. Vrlo često ova komorbidna stanja je teže kontrolisati nego same napade i predstavljaju veliki problem za same pacijente, njihove roditelje ili staraoce ali i ostale članove porodice, što u mnogome utiče na njhov kvalitet života. Pacijenti sa DS u velikom % imaju različit stepen intelektualne nedovoljnosti. Oko 80% pacijenata sa DS doživi adultno doba i u jednoj studiji nastariji pacijent sa DS imao je 60 godina (Catarino et al.2011).
Novu nadu za pacijenate i njihove roditelje, a i lekare koji ih leče, čini Genski tretmani bolesti (Disease Modifying Treatment-DMT). Što se tiče DS u fazi razvoja je nekoliko lekova: STK-001 (antisense oligonucleotide) i EXT -101-adenovirus vektor) za koje se nadamo da će uspeti da obezbede efikasniji tretman DS i omoguće bolji kvalitet života pacijenata sa DS i njihovih porodica.

Prof Dravet i DR Bosanka Jocić Jakubi, na epileptološkom Simpozijumu u Bečićima, CG.
Catarino CB, Liu YW, Liagkouras I et al (2011):Drave syndrome as Epileptic Encephalopathy:Evidence from long term course and Neuropathology. Brain 2011: 134; 2982–3010 doi:10.1093/brain/awr129
Dravet, C., & Oguni, H. (2013). Dravet syndrome (severe myoclonic epilepsy in infancy). Pediatric Neurology Part I, 627–633. doi:10.1016/b978-0-444-52891-9.00065-8
S. Gataullina, O. Dulac, From genotype to phenotype in Dravet disease, Seizure: Eur J Epilepsy (2016), http:// dx.doi.org/10.1016/j.seizure.2016.10.014
Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia. 2011;52(Suppl 2):44-9.
Specchio N, Balestri M, Trivisano M, Japaridze N, Striano P,Carotenuto A, et al. Electroencephalographic features in Dravet syndrome: five-year follow-up study in 22 patients. J Child Neurol. 2012;27:439-44
Wirrell, E.C., Hood, V., Knupp, K.G., Meskis, M.A., Nabbout, R., Scheffer, I.E., Wilmshurst, J., Sullivan, J., 2022. International consensus on diagnosis and management of Dravet syndrome. Epilepsia. doi: 10.1111/epi.17274